Глава 3. Наследственные заболевания углеводного обмена
Все разнообразие углеводов, необходимых человеческому организму, можно распределить на группы:
— моносахариды (глюкоза, галактоза, фруктоза, рибоза, глицериновый альдегид и др.);
— дисахариды (сахароза, лактоза, мальтоза и др.);
— полисахариды (гомополисахариды – гликоген, целлюлоза, крахмал; гетерополисахариды – гликозаминогликаны, протеогликаны, липогликаны).
В настоящее время описаны мутации многих генов, ответственных за синтез ферментов – участников метаболизма углеводов. Частота, распространенность, тяжесть клиники развивающихся при этом заболеваний различны. Остановимся на наиболее изученных и относительно часто встречающихся.
3.1. Моносахаридозы
Галактоземия – наследственное заболевание (аутосомно-рецессивный тип), наблюдается с частотой 1:15-20 тыс.
Выделяют два основных типа:
I тип — недостаточность галактозо-1-фосфат-уридилтрансферазы;
II тип — блок галактокиназы (схема 1).
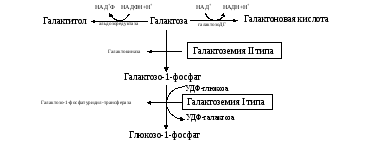
Схема 1. Патология обмена галактозы.
При галактокиназной недостаточности накапливаются галактоза, неиспользованная в синтезе цереброзидов, мукопротеинов, галактотиол – шестиатомный спирт, токсичный для клеток, особенно для нейронов.
При тяжелой форме (I типа) вследствие накопления токсичного галактозо-1-фосфата первые симптомы появляются после прикладывания ребенка к груди. Срыгивания сменяются упорной рвотой, присоединяется диарея, снижается масса тела. Возникает желтуха, увеличиваются печень, селезенка, возможен асцит. В течение нескольких суток развиваются неврологические расстройства, катаракта. При II типе симптоматика менее выражена, но вероятность умственной отсталости, нарушение зрения сохраняются. Диагноз подтверждается регистрацией повышенных концентраций галактозы в крови (в 2 и более раз), а также галактозурией, гипогликемией. Весьма информативно определение содержания галактозо-1-фосфата (увеличено в десятки раз) или активности галактозо-1-фосфат-уридилтрансферазы (значительно снижена) в эритроцитах.
Для предотвращения повреждений нервной системы необходимо полностью исключить молоко с первых дней после рождения и назначать для вскармливания безмолочные смеси, в частности на основе сои и миндаля. Строгую безмолочную диету требуется соблюдать в течение всей жизни. Позднее нужно вводить в рацион рис, растительные масла, яйца, специальные маргарины. Каши следуют готовить на овощных и мясных бульонах. Лекарственная терапия — симптоматическая (антиоксиданты, гепатотропные средства). При необходимости проводят оперативное лечение катаракты.
Фруктоземия – наследственное нарушение обмена фруктозы из-за недостаточности фруктозо-1,6-дифосфатальдолазы, вследствие чего происходит накопление фруктозо-1-фосфата в печени, почках и других тканях (схема 2).
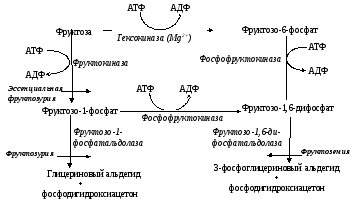
Схема 2. Обмен фруктозы в норме и при патологии.
Первые симптомы обычно появляются у грудного ребенка после введения в питание прикорма (овощных и фруктовых пюре, сладкого чая): возникают рвота, диарея, могут развиться признаки гипогликемии, замедляются рост, прибавка массы тела, возможна гипотрофия.
Тяжелые случаи вследствие гипогликемии могут осложняться потерями сознания, судорогами.
Позднее обнаруживаются гепатомегалия, сопровождающаяся желтухой, в дальнейшем – спленомегалия. Характерно отвращение к сладкой и подслащенной пище. У детей, длительное время находящихся на грудном вскармливании, симптоматика не столь выражена, так как в женском молоке количество моносахаридов меньше, чем в коровьем. Диагностике помогает регистрация в крови гиперфруктоземии, а также повышение активности трансаминаз, щелочной фосфатазы и других ферментов – маркеров функционального состояния гепатоцитов. С помощью реакции Селиванова или методом хроматографии обнаруживают в моче фруктозу (в норме она отсутствует). Диагноз подтверждают выявлением сниженной активности фруктозо-1,6-дифосфатальдолазы в биоптате печени.
Основной принцип лечения – диетотерапия: исключение пищевых продуктов, богатых фруктозой (меда, сахара, сиропов, яблок, груш, арбузов, клубники и др.). Рацион насыщают глюкозой и аскорбиновой кислотой. По мере взросления осторожно вводят фруктозосодержащие пищевые источники.
При строгом соблюдении диеты прогноз вполне благоприятный.
Фруктозурия обычно является бессимптомным нарушением углеводного обмена. Известны две формы данной патологии: одна ограничивается выделением данного моносахарида с мочой без клинических симптомов, другая наряду с фруктозурией характеризуется развитием выраженной гипогликемии при приеме фруктозы и поэтому названа фруктозурией с ее непереносимостью.
Первая патология – эссенциальная фруктозурия обусловлена недостаточной активностью печеночной фруктокиназы, превращающей фруктозу в фруктозо-1-фосфат (схема 2). В этих условиях блокирован обмен кетозы, так как альтернативный путь прямого её превращения в фосфорный эфир с помощью гексокиназы конкурентно подавляется глюкозой, поэтому избыток фруктозы начинает выделяться с мочой.
Фруктозурия с непереносимостью фруктозы. Клинически после приема фруктозы наблюдаются типичные симптомы гипогликемии вплоть до потери сознания и даже смерти. Эта патология обусловлена недостаточностью печеночной фруктозо-1-фосфатальдолазы, что приводит к полному прекращению обмена фруктозы в этом органе. Избыток фруктозо-1-фосфата подавляет активность гликогенфосфорилазы, что в свою очередь, угнетает освобождение глюкозы из гликогена. Фруктозо-1-фосфат ингибирует фруктозо-1,6-дифосфатальдолазу, превращающую глицеральдегид-3-фосфат в фруктозо-1,6-дифосфат и затем в глюкозу (фермент участвует в глюконеогенезе).
Заболевание обычно обнаруживается при переходе на смешанное вскармливание, содержащее сахарозу или фруктозу. После приема пищи наблюдаются рвота, судороги, гипогликемия, позднее – патология печени, почек. Лечение – исключение из диеты фруктозы.
Пентозурия эссенциальная доброкачественная (pentosuria essentialis benigna) — генетический дефект обмена ксилозы, проявляющийся пентозурией, при отсутствии других отклонений; наследуется по аутосомно-рецессивному типу. Данная патология возникает в результате дефицита НАДН-зависимой ксилулозоредуктазы, восстанавливающей L-ксилулозу до ксилитола. Никаких нарушений в состоянии организма, кроме регулярного выведения с мочой нескольких граммов L-ксилулозы, не отмечается.
Источник
61. Наследственные нарушения обмена моносахаридов и дисахаридов: галактоземия, непереносимость фруктозы и дисахаридов, эссенциальная фруктоземия. Гликогенозы и агликогенозы.
Моносахариды — производные многоатомных спиртов, содержащие карбонильную группу. В зависимости от положения в молекуле карбонильной группы моносахариды подразделяют на альдозы и кетозы.
Галактоземия возникает при нарушении обмена галактозы, обусловленном наследственным дефектом любого из трёх ферментов, включающих галактозу в метаболизм глюкозы.
Галактоземия, вызванная недостаточностью галактозо-1-фосфатуридилтрансферазы (ГАЛТ), наиболее хорошо изучена. Это заболевание проявляется очень рано, опасно для детей, так как основным источником углеводов для них служит материнское молоко, содержащее лактозу. Ранние симптомы дефекта ГАЛТ: рвота, диарея, дегидратация, уменьшение массы тела, желтуха. Они появляются вскоре после рождения, как только ребёнок начинает получать молоко. В крови, моче и тканях повышается концентрация галактозы и галактозо-1-фосфата. В тканях глаза (в хрусталике) галактоза восстанавливается альдоредуктазой с образованием галактитола (дульцита). В этой реакции в качестве донора водорода используется NADPH. Восстановление галактозы происходит и в ходе нормального метаболизма, но протекает с небольшой скоростью. При галактоземии галактитол накапливается в стекловидном теле и связывает большое количество воды. Вследствие этого нарушается баланс электролитов, а чрезмерная гидратация хрусталика приводит к развитию катаракты, которая наблюдается уже через несколько дней после рождения.
Тяжёлые последствия дефекта ГАЛТ наблюдают в печени. Это связано с накоплением галактозо-1-фосфата и его токсическим действием на гепатоциты. В результате возникают нарушения функции печени: гепатомегалия, жировая дистрофия. В почках таких больных также повышена концентрация галактитола и галактозо-1-фосфата, что влияет на их функции. Отмечают нарушения в клетках полушарий головного мозга и мозжечка, в тяжёлых случаях — отёк мозга, задержку умственного развития, возможен летальный исход.
Наследственная непереносимость фруктозы, возникающая при генетически обусловленном дефекте фруктозо-1-фосфатальдолазы, не проявляется, пока ребёнок питается грудным молоком, т.е. пока пища не содержит фруктозы. Симптомы возникают, когда в рацион добавляют фрукты, соки, сахарозу. Рвота, боли в животе, диарея, гипогликемия и даже кома и судороги возникают через 30 мин после приёма пищи, содержащей фруктозу. У маленьких детей и подростков, продолжающих принимать фруктозу, развиваются хронические нарушения функций печени и почек. Непереносимость фруктозы — достаточно частая аутосомно-рецессивная форма патологии.
Эссенциальная фруктозурия — редкий, рецессивно наследуемый дефект углеводного обмена. Заболевание связано с недостаточным синтезом фруктокиназы в печени и других тканях.
Непереносимость дисахаридов -наследственная или приобретённая недостаточность активности дисахаридаз, обусловливающая нарушения расщепления и всасывание дисахаридов; вызывает непереносимость лактозы, сахарозы и/или мальтозы; проявляется расстройствами пищеварения и питания в виде хронической ферментативной диспепсии.
Гликогенозы — заболевания, обусловленные дефектом ферментов, участвующих в распаде гликогена. Они проявляются или необычной структурой гликогена, или его избыточным накоплением в печени, сердечной или скелетных мышцах, почках, лёгких и других органах. В таблице 7-3 описаны некоторые типы гликогенозов, различающихся характером и локализацией ферментного дефекта.
Печёночные формы гликогенозов ведут к нарушению использования гликогена для поддержания уровня глюкозы в крови. Поэтому общий симптом для этих форм — гипогликемия в постабсорбтивный период.
Болезнь Гирке (тип I) отмечаютнаиболее часто. Описание основных симптомов этого типа гликогеноза и их причин может служить основанием для понимания симптомов всех остальных типов. Причина этого заболевания — наследственный дефект глюкозо-6-фосфатазы — фермента, обеспечивающего выход глюкозы в кровоток после её высвобождения из гликогена клеток печени. Болезнь Гирке проявляется гипогликемией, гипертриацилглицеролемией (повышением содержания триацилглицеролов), гиперурикемией (повышением содержания мочевой кислоты).
Гипогликемия — следствие нарушения реакции образования свободной глюкозы из глюкозо-6-фосфата. Кроме того, вследствие дефекта глюкозо-6-фосфатазы происходит накопление в клетках печени субстрата — глюкозо-6-фосфата, который вовлекается в процесс катаболизма, где он превращается в пируват и лактат. В крови повышается количество лактата, поэтому возможен ацидоз. В тяжёлых случаях результатом гипогликемии могут быть судороги. Гипогликемия сопровождается уменьшением содержания инсулина и снижением отношения инсулин/глюкагон, что, в свою очередь, ведёт к ускорению липолиза жировой ткани в результате действия глюкагона и выходу в кровь жирных кислот.
Агликогенозы (гликогеноз 0 по классификации) — заболевание, возникающее в результате дефекта гликогенсинтазы. В печени и других тканях больных наблюдают очень низкое содержание гликогена. Это проявляется резко выраженной гипогликемией в постабсорбтивном периоде. Характерный симптом — судороги, проявляющиеся особенно по утрам. Болезнь совместима с жизнью, но больные дети нуждаются в частом кормлении.
Источник